Epigenetic Signatures in Cancer Detection
A case study focused on epigenetic modifications to our DNA and their contribution towards the development and treatment of cancer, with a focus on BRCA1 and MGMT.
This case study is accompanied by a lay summary covering the principles of epigenetics, which can be found here.
Epigenetics is the study of how environmental and heritable modifications can change the expression levels of genes without disrupting the underlying DNA sequence. Changes to the epigenome play a critical role in development, aging, and diseases including cancer. These modifications, including DNA methylation and histone modifications, regulate the structure and accessibility of chromatin to determine whether genes are transcriptionally active or silenced. In cancer, widespread epigenetic dysregulation can promote tumour initiation and influence disease progression. Therefore, an improved understanding of the epigenetic landscape could be a key factor in cancer treatment, enabling more precise tumour classification as well as the identification of vulnerabilities that can be targeted through therapies.
Epigenetic markers include histone modifications and DNA methylation. Eukaryotic chromosomes are made up of repeating protein-DNA complexes called nucleosomes, consisting of a 147 bp DNA sequence wrapped around a histone octamer. This octamer is made up of two copies of histones H2A, H2B, H3 and H4 (Sokolova, Sarkar and Tan, 2023). Epigenetic modifications occur at histone N-terminal tails and influence which proteins can and cannot bind to that specific region. Nucleosomes cluster to form chromatin, which is segregated into active and inactive areas called Topologically Associated Domains, or TADs.
Histone modifications are tags that bind to histone tails in nucleosomes, altering whether the DNA is wound tightly (heterochromatin) or loosely (euchromatin). This, in turn, controls whether genes are transcriptionally active or silenced. In heterochromatin, the DNA is wound tightly around the histone. This prevents transcription factors from binding to the DNA and transcribing genes, essentially switching the unreachable genes off. Alternatively, in euchromatin, the DNA sequence is wound more loosely around the histone. This allows transcription factors to reach and bind to DNA, therefore allowing for genes to be transcribed. These genes are switched on. Two well-known modifications are the addition of methyl and acetyl groups to the histone octamer. Methylation forms heterochromatin whilst demethylation forms euchromatin, and acetylation forms euchromatin whilst deacetylation forms heterochromatin. This process is visualised in Figure 1. Other modifications include phosphorylation, ubiquitination, and biotinylation. Tags can be added to and removed from histones by specific enzymes, rendering epigenetic modifications and their effects on genes reversible. For example, acetyl groups are added to specific lysines by histone acetyl transferases (HATs) and are removed by histone deacetylase complexes (HDACs) (Bozdemir et al., 2025).
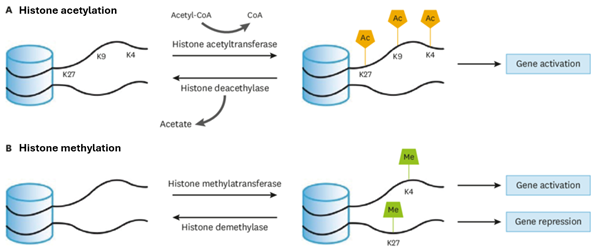
Figure 1: A diagram showing the histone modification processes for acetylation and methylation on N-terminal tails. Image adapted from Lee et al. (2020).
DNA methylation is a dynamic process and an example of a memory system in mammals involving a post-transcriptional replication modification. In sperm and oocytes, DNA is highly methylated, whereas after fertilisation, there is a wave of demethylation followed by a cell-specific methylation process. This is shown in Figure 2.
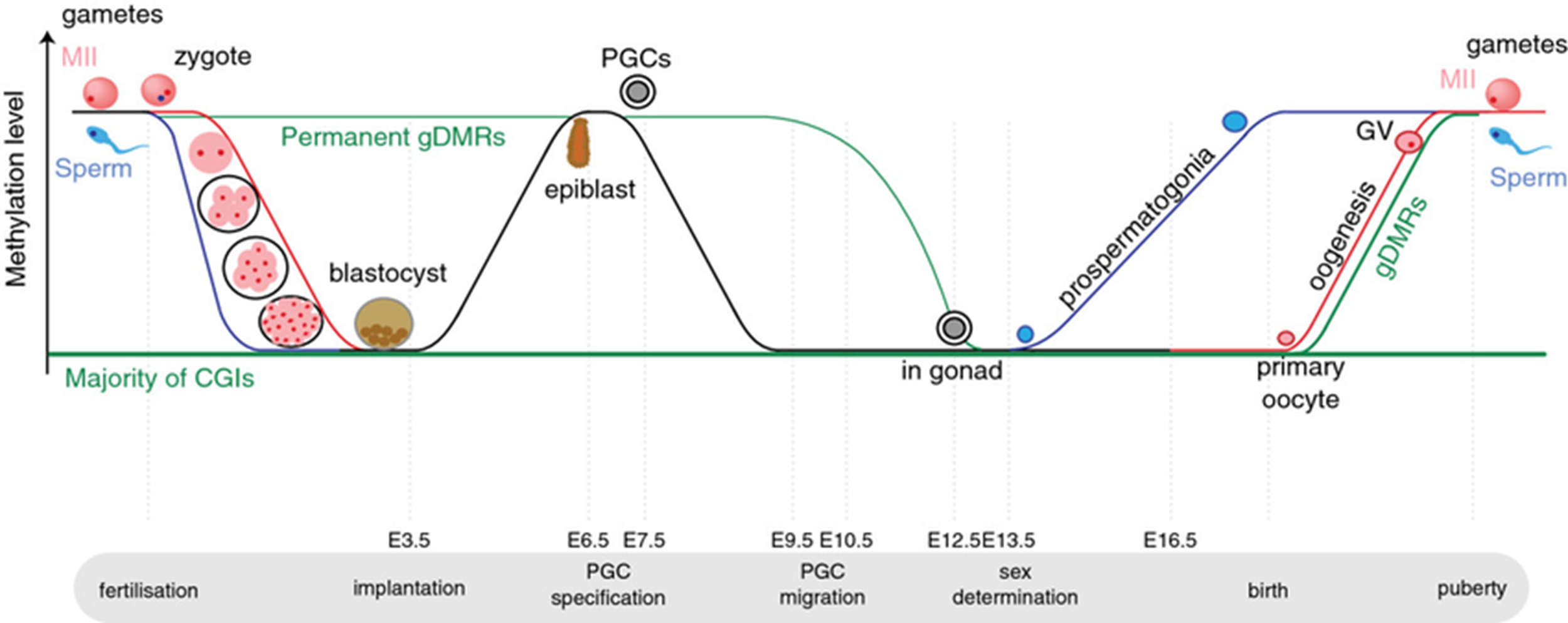
Figure 2: Diagram showing the methylation level of DNA at various stages through early development and fertilisation. Image from Saadeh and Schulz (2014).
CpG dinucleotides are the predominant site of DNA methylation at cytosine bases. De novo methyltransferase enzymes such as DNMT3A and DNMT3B add methyl groups, whilst maintenance enzymes such as DNMT1 copy methyl groups after DNA replication and maintain the pattern, allowing for the epigenetic modification to be ‘inherited’ by daughter cells. S-adenosyl-L-methionine (SAM) is the donor of the methyl group.
In the DNA sequence, there are high density clusters of CpG dinucleotides called CpG islands. These islands are mostly unmethylated despite a high potential for methylation. Promoters are often found within CpG islands. If these islands are unmethylated, genes can be transcribed and are therefore switched on, but methylation of these islands establishes the long-term silencing of a gene due to the promoter being inactive (Saadeh and Schulz, 2014).
In some cases, excessive methyl groups are added to gene promoters, specifically at CpG islands, leading to gene silencing. This is a process in cancer called hypermethylation, often targeting tumour suppressor genes such as BRCA1 (breast cancer 1) in breast and ovarian carcinomas (Oubaddou et al., 2023). The addition of methyl groups to the promoter mimics a loss-of-function mutation (Herman, 1999). The loss of BRCA1 expression compromises DNA repair processes through homologous recombination, which increases the instability of the genome and accelerates tumour progression. By removing a key repair mechanism within DNA, BRCA1 hypermethylation can contribute to the development of cancer.
BRCA1 hypermethylation can also be used as a biomarker for disease progression. Tumours with BRCA1 silencing display an increased sensitivity to PARP inhibitors and DNA-damaging agents including chemotherapy, as the cancer cells are unable to repair DNA due to the hypermethylation process (Oubaddou et al., 2023). This highlights the clinical value of epigenetic profiling alongside genomic sequencing. As epigenetic modifications are reversible, the hypermethylation of CpG islands at BRCA1 promoters is also a target for epigenetic drugs including demethylating agents.
Whilst BRCA1 promoter hypermethylation contributes to tumour progression and genomic instability, the hypermethylation of the MGMT promoter (O^6-methylguanine-DNA methyltransferase) prevents the cell’s ability to repair DNA damage caused by certain chemicals (Rivera et al., 2009). Unlike BRCA1 hypermethylation, MGMT hypermethylation changes the behaviour of cancer once the cells already exist instead of driving cancer progression. As MGMT genes repair DNA damage, keeping the gene silenced allows chemotherapy damage to remain, whilst switching the gene back on allows the chemotherapy damage to be fixed. Therefore, MGMT hypermethylation allows for a better response to treatment therapies including temozolomide (Rivera et al., 2009).
Together, the contrasting roles of BRCA1 and MGMT hypermethylation in cancer development and treatment illustrate a central principle of precision medicine techniques. Cancer treatment is increasingly guided not only by which genes are mutated, but also by which genes are epigenetically altered. The integration of epigenetic profiling alongside genomic data can allow scientists to better predict the behaviour of tumours, identify therapeutic vulnerabilities, and tailor treatment strategies to individual patients rather than relying on uniform protocols (Yu et al., 2024). Ongoing efforts to integrate epigenetic biomarkers into diagnostic workflows, alongside the development of epigenetic therapies, will be critical to personalised cancer care.
References
Bennett, R.L. and Licht, J.D. (2018). Targeting Epigenetics in Cancer. Annual Review of Pharmacology and Toxicology, 58(1), pp.187–207. doi:https://doi.org/10.1146/annurev-pharmtox-010716-105106.
Bozdemir, N., Kablan, T., Biyikli, E., Cinar, O. and Uysal, F. (2025). A comprehensive review of histone modifications during mammalian oogenesis and early embryo development. Histochemistry and Cell Biology, 163(1), pp.70–70. doi:https://doi.org/10.1007/s00418-025-02398-x.
Herman, J.G. (1999). Hypermethylation of tumor suppressor genes in cancer. Seminars in Cancer Biology, 9(5), pp.359–367. doi:https://doi.org/10.1006/scbi.1999.0138.
Lee, H.-T., Oh, S., Ro, D.H., Yoo, H. and Kwon, Y.-W. (2020). The key role of DNA methylation and histone acetylation in epigenetics of atherosclerosis. Journal of Lipid and Atherosclerosis, 9(3), pp.419–434. doi:https://doi.org/10.12997/jla.2020.9.3.419.
Oubaddou, Y., Oukabli, M., Fenniche, S., Elktaibi, A., Elochi, M.R., Al Bouzidi, A., Qmichou, Z., Dakka, N., Diorio, C., Richter, A., Bakri, Y. and Ameziane El Hassani, R. (2023). BRCA1 Promoter Hypermethylation in Malignant Breast Tumors and in the Histologically Normal Adjacent Tissues to the Tumors: Exploring Its Potential as a Biomarker and Its Clinical Significance in a Translational Approach. Genes, [online] 14(9), p.1680. doi:https://doi.org/10.3390/genes14091680.
Rivera, A.L., Pelloski, C.E., Gilbert, M.R., Colman, H., De La Cruz, C., Sulman, E.P., Bekele, B.N. and Aldape, K.D. (2009). MGMT promoter methylation is predictive of response to radiotherapy and prognostic in the absence of adjuvant alkylating chemotherapy for glioblastoma. Neuro-Oncology, 12(2), pp.116–121. doi:https://doi.org/10.1093/neuonc/nop020.
Saadeh, H. and Schulz, R. (2014). Protection of CpG islands against de novo DNA methylation during oogenesis is associated with the recognition site of E2f1 and E2f2. Epigenetics & Chromatin, 7(1). doi:https://doi.org/10.1186/1756-8935-7-26.
Sokolova, V., Sarkar, S. and Tan, D. (2023). Histone variants and chromatin structure, update of advances. Computational and Structural Biotechnology Journal, [online] 21, pp.299–311. doi:https://doi.org/10.1016/j.csbj.2022.12.002.
Yu, X., Zhao, H., Wang, R., Chen, Y., Ouyang, X., Li, W., Sun, Y. and Peng, A. (2024). Cancer epigenetics: from laboratory studies and clinical trials to precision medicine. Cell Death Discovery, [online] 10(1), pp.1–12. doi:https://doi.org/10.1038/s41420-024-01803-z.